 Search
Search
Fetal Urinary System Abnormalities II
Posterior Urethral Valves (PUV), Autosomal Recessive Polycystic Kidney Disease (ARPKD), Cystic Renal Dysplasia, Duplex Kidney and Ureterocele, Renal Agenesis, Horseshoe Kidney
(To view a specific reference. Click on the reference number which will take you to the abstract or article.)
Posterior Urethral Valves (PUV)
Page Links: Definition, Incidence, Antenatal Diagnosis, Embryology and Classification, Fetal Surgical Treatment, Long-term Outcome, Neonatal Diagnosis, Neonatal Treatment,
Definition


Posterior urethral valves are urinary barriers at the level of the posterior urethra near the prostatic urethra that affect male patients. This disorder is among a complex heterogeneous group of urinary abnormalities that include anterior urethral valves, urethral atresia, and bladder neck obstruction, leading to a spectrum of clinical diseases from normal renal function to renal dysplasia and chronic renal disease. Other rare urethral anomalies include: duplication of the urethra, mega-urethra, and prostatic urethral polyps. [1] Severe oligohydramnios may occur before 20 weeks gestation and is associated with pulmonary hypoplasia and perinatal death. Surviving infants have a high likelihood of renal failure leading to dialysis and/or renal transplantation [2], and about 30% develop renal insufficiency before adolescence. [3]
Incidence

PUV is the most common cause of lower urinary obstruction in male infants. PUV occurs in 1:5,000 live births, and in those who survive, 50% progress to end-stage renal disease within 10 years. [4] In some studies, PUV accounts for approximately 13% of prenatally diagnosed cases of hydronephrosis. [5]
Antenatal Diagnosis
The ultrasound characteristics of PUV include: enlarged bladder (megacystis), bilateral ureteral dilatation, bilateral renal pelvis dilatation, oligohydramnios, and potential renal dysplastic changes. [6] The bladder wall is usually thickened and a “keyhole” sign is seen at the junction of the bladder neck. Vesicoureteral reflux is present in approximately two thirds of cases. [7] Urinoma is the extra-renal collection of urine and may be perirenal, urinary ascites, or urinothorax due to renal rupture and occurs in approximately 15% of fetuses with PUV, but renal function may not be different in PUV patients with or without urinoma. [8]
Embryology and Classification

The anatomy and embryology of posterior room urethral valves is not well defined. [9] No proposed theories of valve development are uniformly accepted, although PUV is commonly associated with other urinary anomalies. [10]
Fetal Surgical Treatment

Options for fetal surgery include vesico-amniotic shunting and fetal cystoscopy. Literature surveys indicate that survival and normal renal function occur in 40% to 50% after vesico-amniotic shunting and 65% to 75% after fetal cystoscopy. [11] Vesico-amniotic shunt placement using a double basket catheter is successful in a limited number of patients. [12] Fetal cystoscopy allows views of the upper urethra and may be successful, allowing guide wire passage or hydro- oblation, but technical limitations persist. [13] Overall, prenatal interventions have not resulted in improved outcomes for children with PUV. [14]
Long-term Outcome

The neonatal mortality for PUV has improved due to antenatal diagnosis and early neonatal decompression of the urinary tract. [15] Renal functional impairment correlates with poor bladder compliance, detrusor over-activity, vesicoureteral reflux, and renal dysplasia. [16]

In follow-up of patients 18 or older, chronic renal failure occurs in greater than 50% and greater than one-third of patients who show arterial hypertension, while end-stage renal disease occurs in approximately 20%. [17] By age 31 to 44, 32% of men with PUV are uremic, 21% have moderate renal failure, and bladder dysfunction is present in 40%. [18]
Among data from the national dialysis and transplant registry, PUV accounts for over 35% of patients with end-stage renal disease. [19] Risk factors for progression to end-stage renal disease in children with PUV include a nadir serum creatinine of greater than 1.0 mg/deciliter and severe bladder dysfunction. [20] As expected, among children with congenital anomalies of the kidney and urinary tract (CAKUT), the risk of progression to end-stage renal disease and dialysis is significantly higher in patients with posterior urethral valves who demonstrate renal dysplasia. [21]
Neonatal Diagnosis


Despite the widespread adoption of antenatal ultrasound, over 50% of patients with PUV are not diagnosed until the first year of life. [22] Renal ultrasound is used to assess the degree of hydronephrosis, the presence of bladder wall thickness, and the presence of echogenic and/or dysplastic cystic kidneys. A voiding cystourethrogram provides diagnostic confirmation of antenatal diagnosis of PUV. The ACE gene can be a risk factor in the progression of renal parenchymal damage in children with congenital urinary tract abnormalities. [23] PUV has been associated with decreased renin- angiotensin system activity with ACE II genotype, suggesting that the renin-angiotensin genes may have a role in urinary tract development. [24] Other studies of consanguineous male descendants suggest the possibility of autosomal recessive inheritance of PUV/prune belly syndrome. [25]
Neonatal Treatment

Initial treatment may be catheter drainage after birth. Endoscopic fulguration is the treatment of choice with a demonstrated decrease in mean serum creatinine, hydronephrotic changes, proteinuria, and urinary tract infections. [26] The routine use of high urinary diversion is controversial. [27]
Page Links: Antenatal Diagnosis, Neonatal Diagnosis, References
Antenatal Diagnosis

The ultrasound characteristics of PUV include: enlarged bladder (megacystis), bilateral ureteral dilatation, bilateral renal pelvis dilatation, oligohydramnios, and potential renal dysplastic changes. [28] The bladder wall is usually thickened, and a “keyhole” sign is seen at the junction of the bladder neck. Vesicoureteral reflux is present in approximately two-thirds of the cases. [29] Urinoma is the extra-renal collection of urine and may be perirenal, urinary ascites, or urinothorax due to renal rupture and occurs in approximately 15% of fetuses with PUV. [30]
Neonatal Diagnosis

Despite the widespread adoption of antenatal ultrasound, over 50% of patients with PUV are not diagnosed until the first year of life. [31] Renal ultrasound is used to assess the degree of hydronephrosis, the presence of bladder wall thickness, and the presence of echogenic and/or dysplastic cystic kidneys. A voiding cystourethrogram provides diagnostic confirmation of antenatal diagnosis of PUV.
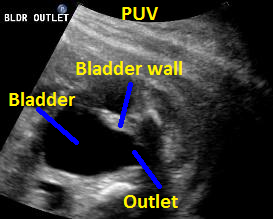
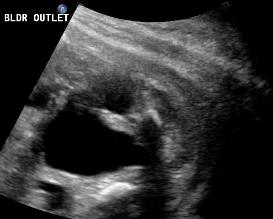
Above. 25 weeks gestation. Posterior urethral valves (PUV) with “keyhole” appearance to the bladder outlet and thickened bladder wall.


Above. Same patient, PUV. Sagittal view demonstrating dilated ureter.
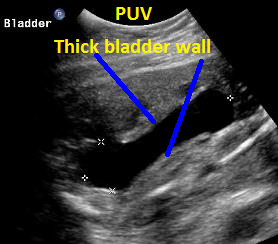

Above. Same patient, PUV. Sagittal view of outlet and and thickened bladder wall.

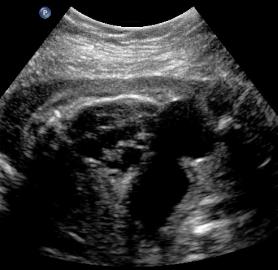
Same patient, PUV. Oblique view with interface identified between the ureter and thickened bladder wall.
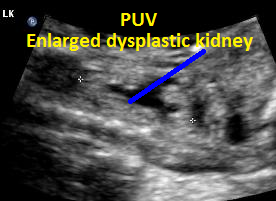

Same patient, PUV. Enlarged dysplastic kidney secondary to PUV outlet obstruction.
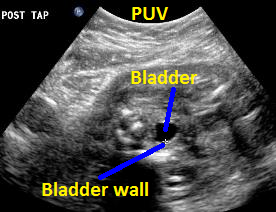

Above. Same patient (25 weeks gestation), PUV. Transverse view of the thickened bladder wall following aspiration of fluid from the fetal bladder.
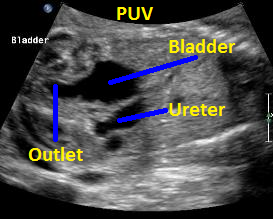

Above. Patient B with PUV. Sagittal view with keyhole appearance of bladder outlet and dilated ureter.

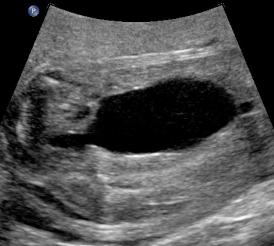
Above. Patient B, PUV. Bladder is more distended and outlet obstruction is more pronounced.
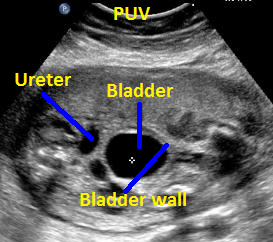
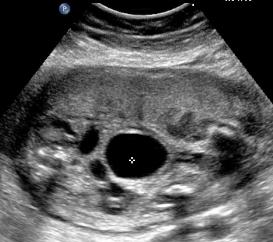
Above. Patient B, PUV. Transverse view demonstrating thickened bladder wall and ureteral dilatation.
Posterior Urethral Valves (PUV).
Above. PUV
Above. PUV
Above. PUV
Above. PUV
Above. PUV
Autosomal Recessive Polycystic Kidney Disease (ARPKD)
Page Links: Definition, Etiology, Incidence, Genetics, Serum Markers, Outcome, Treatment, References
Definition

ARPKD is a severe form of inherited renal disease characterized by dilatation of collecting ducts and hepatic fibrosis. [32] This condition is also known as Infantile Polycystic Kidney Disease and Potters Type 1.
The classic prenatal ultrasound findings include bilateral symmetrically enlarged echogenic kidneys. Oligohydramnios and absent bladder may or may not be a feature.
Etiology

The gene responsible for this disease is (polycystic kidney hepatic disease 1) PKHD1, which is located on chromosome 6, and is expressed in the kidney, pancreas, and liver. The involved proteins, some of which are localized to the cilia of the renal epithelial cells, are the link between ciliary dysfunction and cyst development. [1] Renal cysts are typically small with high water content, accounting for their bright appearance. The expression of the disease is highly variable, and hepatic biliary duct malformations can result in subsequent periportal fibrosis and attendant complications. [33]
Incidence

ARPKD occurs in approximately 1 in 20,000 live births. [1] In women referred for prenatal ultrasound who are at risk or have a family history of urinary tract anomalies, the urinary tract abnormality rate is approximately 8%, but in women with suspected urinary anomalies and with no family history of renal disease, abnormalities of the urinary tract occur in 64%. [34] In another study, ARPKD occurred in 3 of 58 cases of congenital renal malformations. [35] Overall, ARPKD occurs in a minority of patients with fetal urinary malformations. [36] Among lethal urinary tract anomalies, this condition accounts for approximately 14% of cases. [37]
Genetics

Mutations are common since PKHD1 is a large gene on chromosome 6p12. [38] As early as 1998, halo-type prenatal genetic diagnosis was described in pregnancies known to be at risk because of previously affected siblings. [39] Genotype-phenotype correlations have been established to categorize mutations, and the overall detection rate is reported at approximately 77%. [40]
Serum Markers

Increased maternal serum alfa-feto protein (MSAFP) is associated with ARP KD. [41] Although fetal serum ss2– microglobulin and cystatin C are potential predictors of postnatal renal function, they are not useful in the prediction of renal function in ARPKD. [42]
Outcome

The clinical outcome of ARPKD varies greatly, ranging from stillbirth and neonatal death to adult survival. [43] In patients with defined PKHD1 mutations, the following outcomes were observed in 164 cases [44]:
1 year survival: 85%
10 year survival: 82%
Kidney length greater than 97th percentile: 92%
Systemic hypertension: 75%
Congenital hepatic fibrosis and portal hypertension: 44%
Overall molecular detection rate: 76.6%.

In a longitudinal study of 209 patients with ARPKD, the variability of prognosis is as follows [45]:
1. A subset of long-term survivors demonstrates slower progression of the disease than anticipated.
2. Neonatal ventilation is a predictor of mortality but those infants who survive the neonatal period have a better prognosis than originally thought.
3. Disease progress is organ specific between kidney and liver.
4 Only a subset of patients develop periportal hypertension.
The degree of involvement of the kidney and liver tend to be inverse since those with severe renal disease usually have mild hepatic disease. [2]
Treatment

Treatment of survivors is consistent with the above principles [2].
Page Links: Ultrasound Diagnosis (Essential Data), Early Scan, Major Ultrasound Findings, MRI, Differential Diagnosis, References
Ultrasound Diagnosis (Essential Data)

Since there is an overlap in diagnostic criteria among echogenic and cystic renal diseases, the following patient information should be considered: clinical data, family history, general family history of renal abnormalities, other fetal malformations, and fetal syndromes. [46] In addition, in all cases of lethal renal malformations, autopsy is essential since the risk of recurrence is variable between ARPKD (25%) and multi-cystic dysplastic kidney (MCDK) (3%). [47]
Early Scan

A nuchal translucency (NT) of greater than 3 mm. between 9 and 12 weeks may be associated with ARPKD. [48] Echogenic kidneys of normal size have been seen as early as 15 4/7 weeks gestation in patients with ARPKD. [49] Caution is urged when echogenic kidneys are observed in individuals without a history of genetic risk since normal outcome has been reported. [50] In addition, variable outcomes are possible and have been reported among affected fetuses from a single family. [51]
Major Ultrasound Findings

Both clinical outcomes and ultrasound findings are variable in ARPKD. The classic ultrasound findings include bilateral symmetrically enlarged echogenic kidneys with a normal pulsatility index (P-I) of the renal artery. [52] Renal echogenicity is defined as renal parenchymal echogenicity which is greater than that of the liver. Oligohydramnios and lack of fetal bladder distention are additional features. [53] In some cases of ARPKD, normal kidney echogenicity may be present as late as 30 weeks, and oligohydramnios and absent fetal bladder filling may be present in a minority of affected pregnancies. [54] Therefore, oligohydramnios is characteristic of ARPKD but is not always present. [55] However, when present, severe oligohydramnios and renal size are predictors of long-term outcome. Among 14 fetuses no survivors occurred when fetal kidney size was > 4 standard deviations in the presence of severe oligohydramnios. [56] Among 10 infants with oligohydramnios, two of whom had ARPKD, seven of eight children survived and had normal developmental outcomes. [57]

In the presence of echogenic kidneys with normal amniotic fluid volume, both ARPKD and normal kidneys are observed. [58] Among those with normal amniotic fluid volume and renal size of < 4 standard deviations, 14 of 17 infants survived. [11]
In children there may be diffuse renal cystic enlargement without dysplastic findings. [59] Unusual presentations include pyramidal hyper echogenicity which is similar to medullary nephrocalcinosis. [60]
MRI


When sonography is not conclusive, a MRI may be of value. ARPKD is demonstrated as a localized medullary hyperintense lesion. [61] MRI has the potential to reveal enlarged kidneys with low signal density on T1–weighted images and high signal density on T2–weighted images, and these findings are consistent with the high water content in small renal cysts. [62] While ultrasound is superior to MRI for the diagnosis of multicystic dysplastic kidney (MCDK) and uretero-pelvic junction (UPJ) obstruction, MRI is confirmatory for the diagnosis of ARPKD. [63]. In ARPKD, MRI demonstrates clear renal delineation and tissue specificity compared with ultrasound. [64]
Differential Diagnosis

Among 93 fetuses with echogenic kidneys, who later developed nephropathy, cystic characteristics were not always useful for prenatal diagnosis, and only 29% of ARPKD patients exhibited cysts. [65] In that study, echogenic kidneys were seen in the following diagnostic groups:
1. Autosomal dominant and ARPKD.
2. Syndromes which are distinguished by features other than echogenic kidneys:
Bardet-Biedl (obesity, retinitis pigmentosa, polydactylism, hypogonadism, renal failure); Distinguishing ultrasound feature: polydactylsim.
Meckel Gruber (cystic kidneys, occipital encephalocele, and hepatic fibrosis); Distinguishing ultrasound feature: encephalocele.
Ivemark II (asplenia, heart malformations, and situs inversus); Distinguishing ultrasound feature: heart malformations, and situs inversus.
Jarcho-Levin (malformations of the vertebral column and ribs, shortened thorax, and moderate to severe scoliosis and kyphosis); Distinguishing ultrasound feature: thoracic and/or vertebral anomalies.
Beemer (congenital dwarfing skeletal dysplasia); Distinguishing ultrasound feature: thoracic and/or vertebral anomalies.
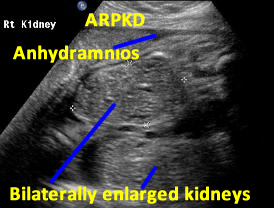

Above. Patient A. Bilateral enlarged kidneys with small cysts and anhydramnios.

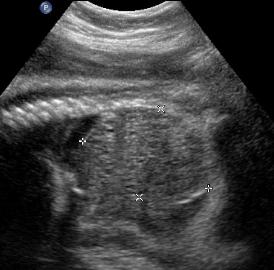
Above. Patient A. Markedly enlarged kidney with small cysts and anhydramnios.
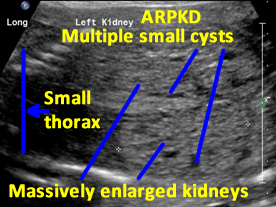
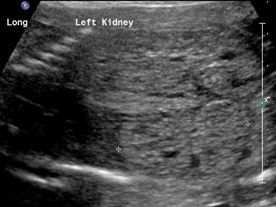
Patient B. Massively enlarged kidneys with small cysts, small thorax and absent amniotic fluid.

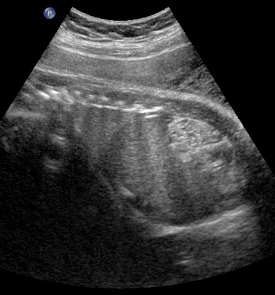
Above. Patient C. 32 2/7 weeks gestation. Bilateral echogenic kidneys with small cysts and oligohydramnios. The renal size is small. Differential diagnosis would include certain syndromes. (See Imaging Considerations, ARPKD).

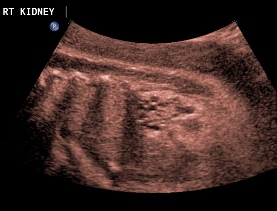
Above. Patient C. 32 2/7 weeks gestation. Bilateral echogenic kidneys with small cysts and oligohydramnios. Outcome likely lethal.
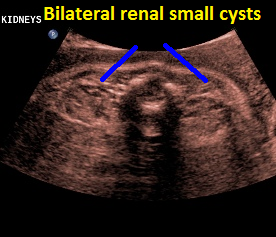
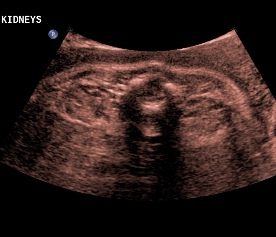
Above. Patient C. 32 2/7 weeks gestation. Transverse. Bilateral echogenic kidneys with small cysts and oligohydramnios.
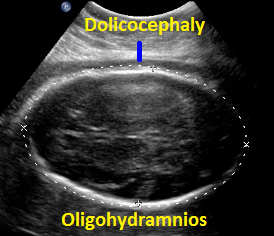

Above. Patient C. 32 2/7 weeks gestation. Dolicocephaly and oligohydramnios.
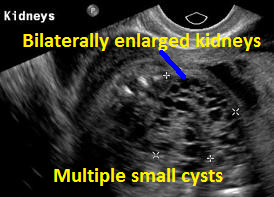

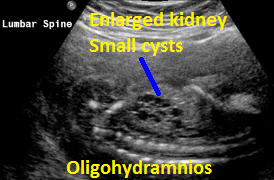
Above Patient D. Larger multiple cysts and enlarged kidneys bilaterally. Differential diagnosis includes bilateral MCDK.
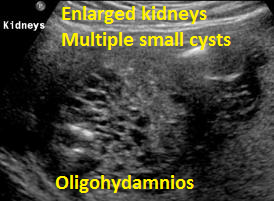
Above Patient D. Larger multiple cysts and enlarged kidneys bilaterally. Outcome, likely lethal.
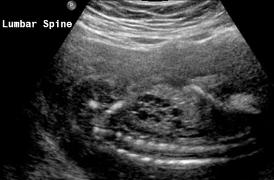
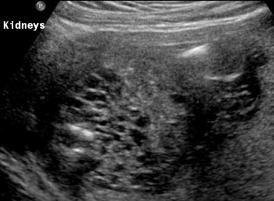
Above Patient D. Larger multiple cysts and enlarged kidneys bilaterally.
Autosomal recessive polycystic kidney disease (ARPKD).
Above: ARPKD. Transverse view. Multiple small cysts.
Above: ARPKD. Longitudinal view: echogenic kidneys
with multiple small cysts.
Back to Top
Cystic Renal Dysplasia


Obstructive renal dysplasia or cystic renal dysplasia is defined as the abnormal development of nephron and duct structures due to urinary tract obstruction. [66] While bilateral cystic renal dysplasia may differ from multicystic dysplastic kidneys, the distinction is often impossible to define with certainty and the prognosis may be similar. Cystic renal dysplasia may either be bilateral or unilateral with contralateral renal abnormalities. Almost half the cases have other congenital anomalies (heart disease, central nervous system, and gastrointestinal malformations). [67]

Cystic renal dysplasia can result from primary dysplasia of the renal parenchyma or from obstruction. [68] Early urinary tract obstruction may cause retention of urine in functioning nephrons and lead to glomerular cysts: expansion of the cysts with tubular dilatation disturbs nephron induction, which in turn may result in obstructive renal dysplasia. [69] Experimentally, a variety of renal dysplastic types have been created following urinary tract obstruction. [70] In the fetal rhesus monkey, obstruction of the urinary tract produces renal dysplasia, which is associated with cell death during early nephron development. [71] Overall, the grade of renal dysplasia is related to fetal pulmonary development. [72] Among patients with Potter’s sequence 47.5% are associated with renal dysplasia. [73]

From an ultrasound perspective, cystic renal dysplasia results in kidneys retaining their renal form. Renal size is normal and/or small and scattered cysts may be smaller than those commonly seen with multicystic dysplastic kidneys. In addition to cysts, increased echogenicity of the renal parenchyma is seen. However, in the newborn, diffusely hyper-echogenic kidneys are seen in a variety of conditions, sometimes requiring renal biopsy to differentiate certain forms. [74]
Some authors distinguish cystic renal dysplasia from glomerulocystic kidney disease (GCKD) with the later categorized into non-syndromic, heritable (dominant), and sporadic forms. Syndromic forms would include Meckel syndrome and heritable malformations would include tuberous sclerosis and trisomy 13. [75]
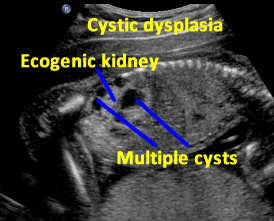
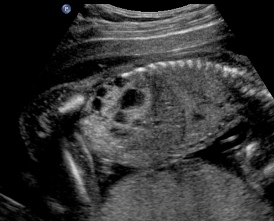
Above. Patient A. Sagittal view. Multiple cysts and echogenic parenchyma are noted. These findings are also seen in multi-cystic dysplastic kidney and the two are difficult to distinguish.
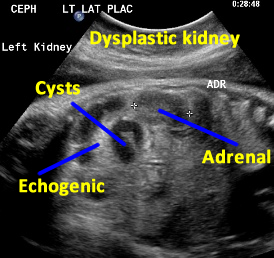

Patient B. Again cysts and echogenic tissue are observed.
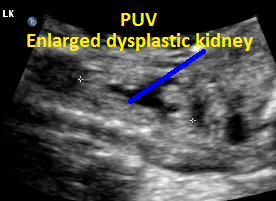
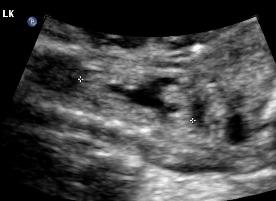
Above. Patient C. This dysplastic echogenic kidney’s findings are secondary to posterior urethral valves.
Duplex Kidney and Ureterocele
Page Links: Development, Definition, Classification, Incidence, Associations and Outcomes, Ureterocele, Pathogenesis, Management: Ureterocele, Management: Ureteral Re-Implantation, Management: Hemi-Nephrectomy, Management: Laparoscopic Hemi-Nephrectomy, References
Development
Around the 4th week of embryonic development, the ureteric bud arises from the mesonephric (Wolffian) duct and gives rise to the ureter and portions of the urinary collecting system. In a duplicated collecting system there is division into an upper and lower pole of the kidney. [76]
Definition

Duplicated collecting system or duplex kidney is defined as a division of the renal collecting system into separate upper and lower poles.
According to the American Academy of Pediatrics [77] a duplex kidney is defined as a kidney with 2 pelvic caliceal systems.
Classification

Partial duplication: includes those with a single ureter or with bifid ureters that join before emptying into the bladder.
Complete duplication: includes duplex kidneys with 2 ureters that empty separately into the genital or urinary tract.
Incidence
In in large series of patients, 2.3% had duplex kidneys and 83.2% were unilateral and 16.8% bilateral. The finding was equally common on the right and left side but was twice as common in females compared to males. [78]
Associations and Outcomes

Evidence of renal disease was seen in 27% of the duplex kidneys and 3% of non-duplex kidneys with the most common consequence being urinary reflux. [3]
In another series with high grade primary vesicoureteral reflux, duplex kidney was the main associated anomaly occurring in 148 (64.6%) of 229 patients. [79]
Duplex collecting systems were noted in 8% of girls referred for urinary tract infections. [80] In 69% of complete duplex and in 22% of partial duplex systems, vesicoureteral reflux was observed while scarring was noted in in 17% of complete duplication and 6% of incomplete duplication. Those with incomplete or partial duplication had similar outcomes compared to those with non-duplex kidneys. [5]
Ureterocele
Although variously reported as one of the associations with duplex kidney, a ureterocele is a saccular expansion of the distal ureter. (bladder implantation site). Commonly seen in females and children it usually affects the upper pole of the duplication. [81]
Four types of ureteroceles are described [6]:
(A) ureterocele with single ureter (10%);
(B) ureterocele with total duplication and intravesical development (10%);
(C) ureterocele with total duplication and extravesical development (62%); and
(D) ureterocele with ectopic ureter (3%).
Kidneys with a duplicated collecting system are larger than the contralateral kidney and the duplex kidney contributes 51 to 67% to total renal function compared to the opposite kidney which contributes 33 to 49%. [82]
Pathogenesis
Recognizing that congenital anomalies of the kidney and urinary tract (CAKUT) are frequent and are related to chronic renal disease, a genetic contribution is suspected, and a consortium is established to study the molecular and genetic etiology of these conditions. [83]
Management: Ureterocele

The management of ureteroceles is variable among urologists and depends upon neonatal age and findings. The following are 233 survey responses from urologists [84]:
1. Intravesical ureterocele with poor upper pole function: Ureterocele puncture at age 3 months (50.6%);
2. Ureterocele, conservative follow up for 18 months, followed by partial nephrectomy (61.8%);
3. Ureterocele without hydronephrosis:
a. Observation (47.2%)
b. Ureterocele puncture (35.6)
c. 16.3% chose partial nephrectomy (16.3%).
The treatment of ectopic ureterocele associated with duplex system is primary endoscopic incision. This is reported as a safe and effective procedure with initial decompression achieved in 93%. [85] Transurethral ureterocele puncture is less likely to result in the need for future surgery in patients with a single ureteral implantation site and/or an intravesical ureterocele. [86]
Prenatal diagnosis of duplex kidneys results in earlier treatment of ureteroceles and reduces the long term morbidity from urinary tract infections and from additional surgery. [87]
Management: Ureteral Re-Implantation

When the ureter implants ectopically, re-implantation into to the bladder results in minimal bladder ureteral reflux. [88]
Management: Hemi-Nephrectomy
Hemi-nephrectomy for duplex kidney results in a significant decrease in renal function in 8% of patients and a small decrease in function in the remaining moiety in 51%. [89]
Management: Laparoscopic Hemi-Nephrectomy
In the pediatric population, laparoscopic hemi-nephrectomy for duplex kidney is pursued by a number of institutions and results in satisfactory outcomes with a non-functioning renal moiety rate of 5%. [90] In addition, a number of other laparoscopic approaches to the management of duplex kidneys are reported. [91],[92]
Page Links: Imaging Considerations, Prenatal Ultrasound Findings, MRI, References
Imaging Considerations
The role of ultrasound in the diagnosis of duplex collecting systems recognized the importance of a dilated upper pole, ectopic ureter and ureterocele. [93] In assessing the role of hydronephrosis in prenatal ultrasound, duplex kidneys account for 5.4% of fetal hydronephrosis. [94]
Prenatal Ultrasound Findings

Early reports suggested the following findings for duplex kidneys [95]:
- Sagittal length above the 95th centile for gestational age,
- Cystic structure in the upper pole,
- Ureterocele (cyst like structure) in the urinary bladder.

In another series of 11 prenatal patients, the median age at diagnosis was 28 weeks and the following ultrasound findings were recorded [96]:
- Hydronephrosis of upper pole (10/11, 91%)
- Mega-ureter (7/11, 64%)
- Ureterocele (6/11, 55%)

False positive diagnosis included: ureteropelvic junction obstruction in a malrotated kidney and obstructed mega-ureter while a false diagnosis included severe hydronephrosis. [4]
Other associated anomalies in girls with duplex collecting systems include: malrotation, ureteropelvic junction obstruction, and bladder diverticulum. [97]
Among 21 confirmed duplex kidneys, two separate renal pelvises was the most common prenatal sonographic feature (n = 15/21 [71%]), followed by dilatation of a single moiety with a dilated same sided ureter and/or findings of a ureterocele (n = 6/21 [29%]). [98] The accuracy of the prenatal diagnosis of duplex kidneys is reported as 75%. [99]
MRI
MRI has been used successfully in the diagnosis of renal abnormalities in in the fetus [100] and neonatal MRI is useful in defining poorly functioning moieties of the duplex kidney. [101]
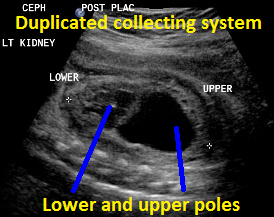

Above. Duplicated collecting system (DCS), 29 weeks gestation. Sagittal view. Note upper pole and lower pole with greater dilatation of the upper pole.
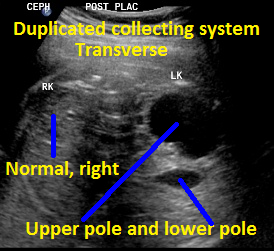
Above. Same patient, DCS. Transverse view with left kidney suggesting DCS with potential upper and lower poles.


Same patient, DCS. Sagittal view of contralateral (right) kidney, which is normal.


Above. Same patient, DCS. Note dilated ureter exiting from upper pole.
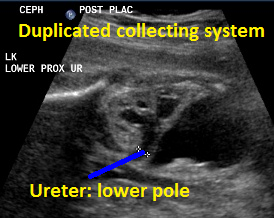
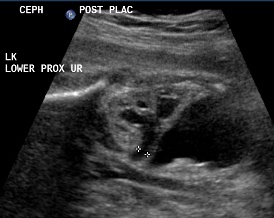
Above. Same patient, DCS. Similarly, less dilated ureter exits from lower pole.


Above. Same patient, DCS. Note fetal bladder and classic appearance of ureterocle, likely representing the distal end of the ureter exiting from the upper pole.


Above. Same patient, DCS. Transverse to oblique view of ureterocele.
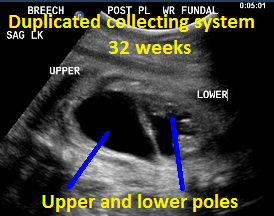
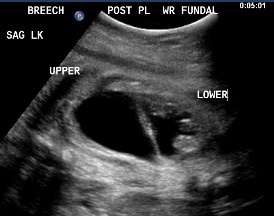
Above. Patient B, DCS. 32 weeks gestation. Note dilated upper and lower poles of the kidney.
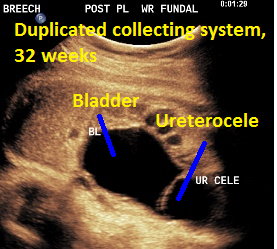

Patient B, DCS. Classic view of ureterocele suggesting the presence of DCS.
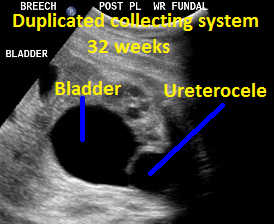
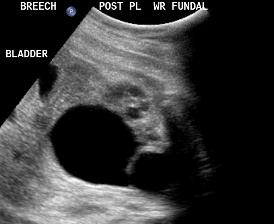
Patient B, DCS. Similar view of ureterocele suggesting the presence of DCS.
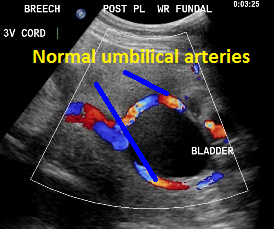

Umbilical arteries around the fetal bladder.
Duplex Kidney and Ureterocele.
Above: Duplex kidney and ureterocele
Back to Top
Renal Agenesis
Page Links: Definition, Unilateral Renal Agenesis, Bilateral Renal Agenesis: Incidence, Pathogenesis, Etiology, Risk Factors, Associated Malformations, Management, References
Definition
Renal agenesis is the absence of one or of both kidneys.
Unilateral Renal Agenesis

Unilateral renal agenesis can be identified during routine fetal ultrasound but requires close observation of both renal fossae. Unilateral renal agenesis may be isolated or associated with chromosomal or non-chromosomal syndromes including malformations of Müllerian structures, association with VACTERL, and association with congenital heart defect. [102] Some cases represent the end-stage involution of a multicystic dysplastic kidney. [103]
Bilateral Renal Agenesis: Incidence

Bilateral renal agenesis occurs in approximately 1.3 per 10,000 live births with a male to female ratio of 2.5:1. [104] In 1946, Edith Potter described the facial characteristics of infants with renal agenesis; these were later found to be due to a deformation caused by lack of amniotic fluid. Overall, the incidence of urinary tract anomalies is 0.9%. [105],[106] Among 117 cases of renal malformations, renal agenesis was identified in 12.8%. [107]
Pathogenesis

During the fifth week of gestation, bilateral ureteric buds arise, growing toward the head from the sacral region until there is contact with the metanephric blastema. [108] When the ureteric bud fails to form, renal agenesis occurs. Fetal urine is initially produced during the tenth week of gestation, and by the fifteenth week, the major component of amniotic fluid is urine, which plays a role in amniotic fluid metabolism. [109] In bilateral renal agenesis, no urine production is possible, and severe oligohydramnios occurs. Fetal lung hypoplasia results and early neonatal death is uniform.
Etiology

Genetic factors are important etiological components. Bilateral renal agenesis may be transmitted in a polygenic pattern fashion. [110] In patients with bilateral renal agenesis, 14.7% had relatives with the same diagnosis compared to the control frequency of 2.2%. For an individual with bilateral renal agenesis, the risk for a first-degree relative is 6.2%. [111] Family histories suggest autosomal dominant genes with incomplete penetrance in non-syndromic bilateral renal agenesis and cystic dysplasia. [112]
In summary, autosomal dominant conditions with incomplete penetrance, complete non-penetrance, and single gene syndromic disorders have been reported. [113]
Risk Factors

A body mass index of greater than 30 kg/meter, pre-conceptual smoking, and binge drinking during the second month of pregnancy are associated with an increased risk for renal agenesis or hypoplasia. [114]
Associated Malformations

Other congenital defects are reported in patients with bilateral renal agenesis. [115] In post-mortem studies of bilateral renal agenesis, and/or hypoplasia, and/or dysplasia, almost 69% were associated with extra-renal malformations, which had a male to female ratio of 2.44:1. [116] In that study, 80 different malformations were reported and included: undescended testes, anorectal atresia, vertebral malformations, incomplete lobulation of the lung, malrotation of the gut, and radial aplasia. In addition, there is a strong association with VACTERAL (vertebral malformation, anal atresia cardiovascular anomalies, TE fistula, esophageal atresia, renal anomalies, and limb malformations). Further, bilateral renal agenesis alone was associated with extra-renal abnormalities in about 36% of cases and also overlapped with VACTERL, caudal dysgenesis, hemifacial microsomia, and Müllerian abnormalities.
Management


In unilateral agenesis, vesicoureteral reflux is the most common contralateral renal abnormality, and while the overall prognosis is generally good, hypertension and renal insufficiency may develop in some patients. Long-term studies of patients with a single kidney suggest the need for lifetime follow-up. [117] Bilateral renal agenesis with anhydramnios is fatal. In the multi-national European registry, terminations of pregnancy were performed in 67% of the detected bilateral renal agenesis/dysgenesis patients and in 4% of unilateral multi-cystic dysplastic kidneys. [118]
Page Links: Unilateral Renal Agenesis, Bilateral Renal Agenesis, References
Unilateral Renal Agenesis


Careful consideration of each renal fossa is necessary to identify unilateral renal agenesis. The empty renal fossa is often an isolated finding but may be also associated with other non-renal malformations. [119] In unilateral renal agenesis, cardiac malformations are the most common non-renal anomaly, but associations are reported with VACTERL*, chromosome abnormalities, and other non-chromosomal syndromes. [120] In unilateral renal agenesis, the contralateral kidney is commonly enlarged with the mean renal length > 95th percentile for gestational age. [121] MRI is a complimentary imaging modality in renal malformations including renal agenesis. [122]
In the absence of the kidney, on the coronal scan, the adrenal gland has a flattened appearance.
Color flow Doppler usually demonstrates the absence of the renal artery.
If the fetus is in breech presentation, transvaginal ultrasound may be useful.
Summary of sonographer tasks in suspected unilateral renal agenesis:
- Use transvaginal probe if fetus is in breech presentation.
- Look for renal tissue in the fetal pelvis.
- Identify the adrenal gland in the coronal plane of the renal fossa.
- Identify the fetal bladder.
- Assess amniotic fluid index (usually normal).
- Use color flow Doppler in the coronal plane to assess for the presence of the renal artery.
- Measure the mean renal length in the contralateral kidney (usually > 95th percentile).
- Search for non-renal malformations, especially cardiac.
- MRI may be a complementary modality in questionable cases.
Bilateral Renal Agenesis
In bilateral renal agenesis, the lack of amniotic fluid creates imaging challenges.
Summary of sonographer tasks in suspected bilateral renal agenesis:
- Use transvaginal probe if fetus is in breech presentation.
- Identify the adrenal gland in the coronal plane of the renal fossa.
- Fetal bladder is non-visualized.
- Use color flow Doppler to identify the umbilical artery or arteries in the fetal pelvis.
- Assess amniotic fluid index (extreme oligohydramnios or anhydramnios after 15 weeks).
- Use color flow Doppler in the coronal plane to assess for the presence of the renal arteries (usually absent).
- Search for non-renal malformations; VACTERL* and cardiac are especially common.
- Assess for pulmonary hypoplasia (chest/trunk length ratio [123] or cardiac to chest circumference ratio).
- MRI may be a complementary modality.
* Vertebral malformation, anal atresia cardiovascular anomalies, TE fistula, esophageal atresia, renal anomalies, and limb malformations
Unilateral Renal Agenesis

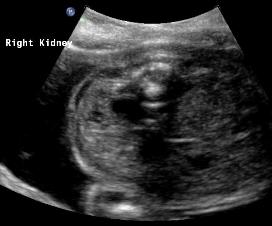 Above: 25 3/7 weeks gestation. Unilateral renal agenesis with dilated renal pelvis on the right. Left renal fossa area, probable adrenal gland. Amniotic fluid volume is normal.
Above: 25 3/7 weeks gestation. Unilateral renal agenesis with dilated renal pelvis on the right. Left renal fossa area, probable adrenal gland. Amniotic fluid volume is normal.

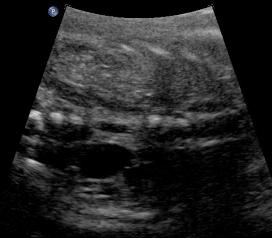 Above. Same patient. Unilateral renal agenesis. Coronal view with right kidney and left renal fossa seen with probable adrenal gland.
Above. Same patient. Unilateral renal agenesis. Coronal view with right kidney and left renal fossa seen with probable adrenal gland.
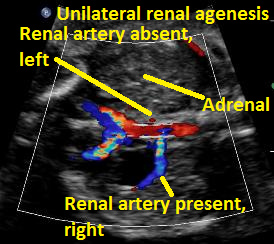
 Above. Same patient Unilateral renal agenesis. Color Doppler demonstrates present of right renal artery only.
Above. Same patient Unilateral renal agenesis. Color Doppler demonstrates present of right renal artery only.
Bilateral Renal Agenesis


Above: A. 19 1/7 weeks gestation. Coronal view. Empty renal fossa with adrenal gland seen. Virtually no amniotic fluid identified.
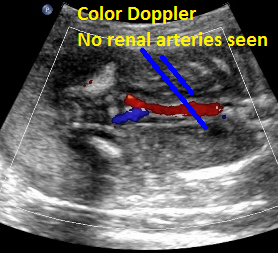
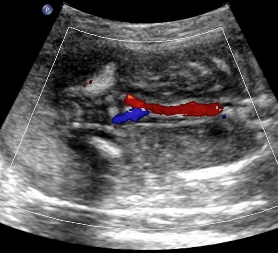
Above: same patient. Color Doppler demonstrates the absence of renal arteries bilaterally.
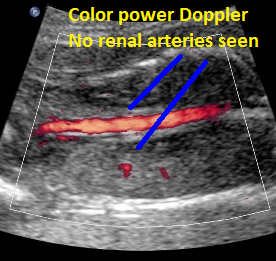

Above: same patient. Color power Doppler demonstrates the absence of renal arteries bilaterally.
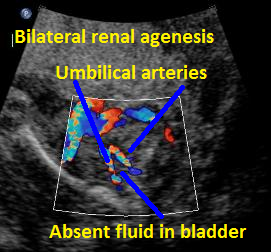
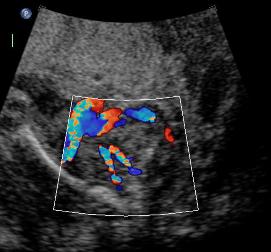
Above B. 18 3/7 weeks gestation. Bilateral umbilical arteries are seen in their normal position but there is no fluid within the bladder.
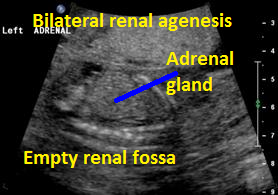
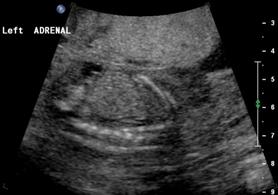
Above. Same patient B. Again, empty renal fossa with adrenal gland visualized.

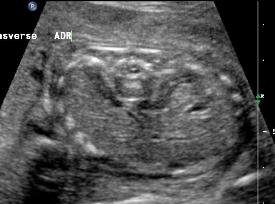
Above C. 22 6/7 weeks gestation. Transverse view. Bilateral empty renal fossae with a portion of the adrenal seen. Amniotic fluid is absent.
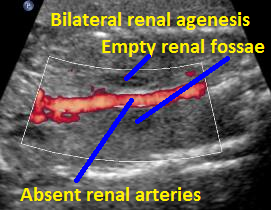

Above C. 22 6/7 weeks gestation. Color power Doppler demonstrating absent renal arteries. Coronal view with empty renal fossae.
Horseshoe Kidney, Pelvic Kidney, Crossed Fused Renal Ectopia
Page Links: Definitions, Empty Renal Fossa, Horseshoe Kidney, Color Flow Doppler, Pelvic Kidney, Crossed Fused Renal Ectopia, References

In the case of empty renal fossa, an ectopic location of the kidney as well as associated malformations must be sought. A unilateral empty renal fossa suggests the possibility of: horseshoe kidney, pelvic kidney, crossed fused renal ectopia, or renal agenesis. [124]
Definitions
In horseshoe kidney, the kidneys are horseshoe shaped and meet in the midline at the isthmus. In pelvic kidney, the kidney is located in the pelvis. In crossed fused renal ectopia, the kidneys are usually fused on one side.
Empty Renal Fossa

In a prospective study of 17 fetuses with empty renal fossa, there were 9 cases of ectopic kidney, 7 cases of pelvic kidney, 1 case of iliac kidney, and 1 case of crossed fused ectopia. [125]

In this study, one case was associated with a cardiac malformation and single umbilical artery while 16 of the 17 cases had good outcomes. In the presence of an empty renal fossa, an ectopic location is noted in 42% (34 of 39 cases in the pelvis and 4 fused to the contralateral kidney), while congenital absence of the kidney is noted in 47%. [126] In this study, 42% of the patients demonstrated other anomalies, including genitourinary tract and cardiovascular anomalies. In other studies of empty renal fossae, 24 of 40 cases are associated with pelvic kidney, and 13 of 40 cases are associated with renal agenesis; 2 of 40 are associated with horseshoe kidney. [127] In this study, the prevalence of empty renal fossa is approximately 3.2 per 1,000, and the overall prognosis is good.
Horseshoe Kidney

Above are sonographic features and findings specific to the antenatal diagnosis of horseshoe kidney. [128]
Among 19 fetuses with horseshoe kidney, 15 had no other abnormalities. Four of 19 fetuses had severe complex abnormalities, three of which were associated with trisomy 18. [129]
Color Flow Doppler

In cases of renal agenesis, the renal arteries are not always seen with color Doppler. [130] Horseshoe kidney may be associated with high bifurcation of the abdominal aorta with aberrant renal artery blood flow to the affected kidney. [131] In renal malformations, variations of the renal vessels are commonly found, and in horseshoe kidney and in pelvic kidney, the renal artery may originate from the iliac artery. [132] Therefore, an aberrant renal artery arising from the iliac artery is commonly a sign of fetal horseshoe kidney.
Pelvic Kidney
In the presence of an empty renal fossa, a search for a pelvic kidney should be undertaken and is sometimes found near the bladder or the fetal bony pelvis.
Isolated unilateral renal agenesis and a fetal pelvic kidney carry limited risks. [133] The prevalence of pelvic kidneys is reported as 0.14%. [134] In this study of 36 newborns with confirmed pelvic kidneys, 33% had some impairment of renal function, but overall renal function remained normal. Among 31 infants available for follow-up only, one case required nephrectomy due to multicystic dysplastic kidney.
Crossed Fused Renal Ectopia
Crossed fused renal ectopia is less commonly found and most often involves fusion of the kidneys, which are enlarged, bilobed, and may be obstructed; the kidney is located on the opposite side of the ureter. [135]
Horseshoe Kidney
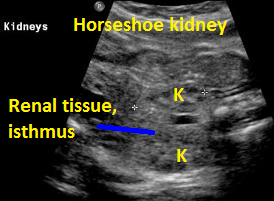

Above. Horsehoe kidney. Coronal view. Elements of renal tissue join in the mid-line.
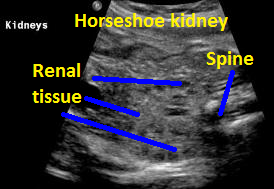
Above. Same patient. Coronal view. Definitive renal tissue can be seen in the mid-line (isthmus).

Above. Kidneys joining in the mid-line with definitive “horseshoe” shape.
Pelvic Kidney


Above: 20.1 weeks gestation. Unilateral renal agenesis with pelvic kidney. Echogenic tissue immediately inferior to empty fossa likely adrenal gland.
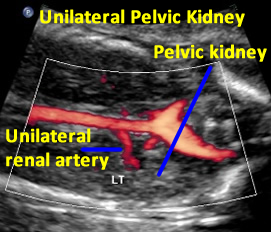

Above. Same patient demonstrating color power Doppler blood flow with a single renal artery to the pelvic kidney.

Same patient. Transverse view of the pelvis at the level of the bladder demonstrating pelvic kidney adjacent to the bladder.
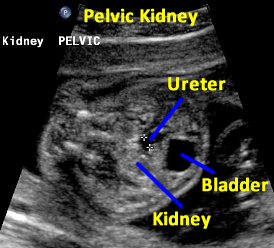
Same patient. Transverse view of the pelvis at the level of the bladder demonstrating ureter and the pelvic kidney adjacent to the bladder.
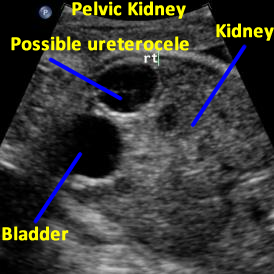
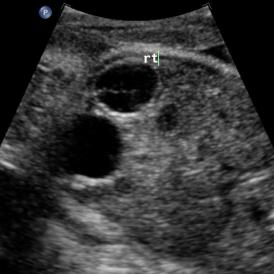
Above: 29 5/7 weeks gestation. Right pelvic kidney adjacent to the bladder with cystic structure, possible ureterocele.


Above. Patient with enlarged kidney on the left side extending to the pelvis and solitary kidney on the right side.


Above. Same patient. Color flow Doppler indicates two renal arteries to the left kidney and one renal artery to the right kidney. The left kidneys extend to the plevis. A renal pelvis is identified in two segments on the left kidney.
Crossed renal ectopia


Above. 29 weeks gestation. Renal ectopia with enlarged kidney extending to the pelvis; the contralateral kidney is absent.
References
1-
Abstract: PMID: 17602414
2-
Abstract: PMID: 21839591
3-
Abstract: PMID: 19838598
4-
Abstract: PMID: 21860792
5-
Abstract: PMID: 19237812
6-
Abstract: PMID: 15604570
7-
Abstract: PMID: 22046789
8-
Abstract: PMID: 18710723
9-
Abstract: PMID: 16515962
10-
Abstract: PMID: 19838598
11-
Abstract: PMID: 21413041
12-
Abstract: PMID: 16817090
13-
Abstract: PMID: 14691987
14-
Abstract: PMID: 15558286
15-
Abstract: PMID: 19838598
16-
Abstract: PMID: 15126863
17-
Abstract: PMID: 21748651
18-
Abstract: PMID: 16094041
19-
Abstract: PMID: 19373494
20-
Abstract: PMID: 19833558
21-
Abstract: PMID: 19536081
22-
Abstract: PMID: 19838598
23-
Abstract: PMID: 15470205
24-
Abstract: PMID: 16006956
25-
Abstract: PMID: 15912376
26-
Abstract: PMID: 19623154
27-
Abstract: PMID: 19838598
28-
Abstract: PMID: 15604570
29-
Abstract: PMID: 22046789
30-
Abstract: PMID: 18710723
31-
Abstract: PMID: 19838598
32-
Abstract: PMID: 14741187
33-
Abstract: PMID: 10835131
34-
Abstract: PMID: 3153330
35-
Abstract: PMID: 16174570
36-
Abstract: PMID: 1984303
37Boutheina BR, Aïda M, Lamia S, Ali M, MedBadis C, Samy J, Issam L, Ezzeddine S, et al. Lethal uropathies: prenatal diagnosis and feto-pathologic aspects. Tunis Med. 2000 Feb;78(2):120-4.
Abstract: PMID: 10894048
38-
Abstract: PMID: 15108277
39-
Abstract: PMID: 9511976
40-
Abstract: PMID: 15698423
41-
Abstract: PMID: 2453819
42-
Abstract: PMID: 15164404
43-
Abstract: PMID: 15108281
44-
Abstract: PMID: 15698423
45-
Abstract: PMID: 12728091
46-
Abstract: PMID: 1781072
47-
Abstract: PMID: 18596710
48-
Abstract: PMID: 9166801
49-
Abstract: PMID: 8559760
50-
Abstract: PMID: 1606120
51-
Abstract: PMID: 1536230
52-
Abstract: PMID: 10401236
53-
Abstract: PMID: 1785280
54-
Abstract: PMID: 2204162
55-
Abstract: PMID: 3287366
56-
Abstract: PMID: 12504976
57-
Abstract: PMID: 11770813
58-
Abstract: PMID: 16032764
59Khoory BJ, Fanos V. Renal polycystosis in pediatrics. Pediatr Med Chir. 1998 Jul-Aug;20(4):269-73.
Abstract: PMID: 9866850
60-
Abstract: PMID: 16491512
61-
Abstract: PMID: 14975971
62-
Abstract: PMID: 9564103
63-
Abstract: PMID: 9197441
64-
Abstract: PMID: 8542471
65-
Abstract: PMID: 17094077
66-
Abstract: PMID: 1870925
67-
Abstract: PMID: 1870925
68-
Abstract: PMID: 12386283
69-
Abstract: PMID: 12386283
70-
Abstract: PMID: 11685705
71-
Abstract: PMID: 11168926
72Shimada K, Hosokawa S, Matsumoto F, Matsumoto S. Pathological study of the kidney and the lung in patients with Potter sequence. Nihon Hinyokika Gakkai Zasshi. 1997 Jul;88(7):664-9.
Abstract: PMID: 9267130
73-
Abstract: PMID: 6393764
74-
Abstract: PMID: 8518103
75-
Abstract: PMID: 12432113
76http://en.wikipedia.org/wiki/Duplicated_ureter
77-
Abstract: PMID: 6502807
78-
Abstract: PMID: 1000896
79-
Abstract: PMID: 21994077
80-
Abstract: PMID: 3492875
81-
Abstract: PMID: 20854476
82-
Abstract: PMID: 14734907
83-
Abstract: PMID: 22121240
84-
Abstract: PMID: 20728105
85-
Abstract: PMID: 21168903
86-
Abstract: PMID: 20728127
87-
Abstract: PMID: 15758799
88-
Abstract: PMID: 22398644
89-
Abstract: PMID: 15821573
90-
Abstract: PMID: 21527211
91-
Abstract: PMID: 21256545
92-
Abstract: PMID: 21437700
93-
Abstract: PMID: 409121
94-
Abstract: PMID: 14676444
95-
Abstract: PMID: 8705408
96-
Abstract: PMID: 10400048
97-
Abstract: PMID: 3492875
98-
Abstract: PMID: 21632996
99-
Abstract: PMID: 12704741
100-
Abstract: PMID: 12386558
101-
Abstract: PMID: 21897576
102-
Abstract: PMID: 8540439
103-
Abstract: PMID: 8201519
104-
Abstract: PMID: 20811621
105-
Abstract: PMID: 9037946
106-
Abstract: PMID: 21536809
107-
Abstract: PMID: 16116932
108O’Rahilly R, Muller, F. Human Embryology and Teratology, Wiley Liss, NY, 1996, pp. 279-279.
109-
Abstract: PMID: 16174570
110-
Abstract: PMID: 7877959
111-
Abstract: PMID: 16977473
112-
Abstract: PMID: 6393764
113-
Abstract: PMID: 20811621
114-
Abstract: PMID: 18835865
115-
Abstract: PMID: 1814304
116-
Abstract: PMID: 20811621
117-
Abstract: PMID: 8540439
118-
Abstract: PMID: 16053904
119-
Abstract: PMID: 22581658
120-
Abstract: PMID: 8540439
121-
Abstract: PMID: 22581658
122-
Abstract: PMID: 20618240
123Abourachid H, Dahmani F, Louis D, Hakami F, Daher N. Etiology of stenoses of the urethra. II–After trans-vesical prostatectomy. Apropos of 2 comparative series. Review of the literature. J Urol (Paris) 1989;95(7):412-4.Abstract: PMID: 2687395
124-
Abstract: PMID: 21084958
125Markov D, Atanassova D, Pavlova E, Markov P. Empty renal fossa–a prenatal diagnostic dilemma. Akush Ginekol (Sofiia). 2010;49(5):13-9.
Abstract: PMID: 21268397
126-
Abstract: PMID: 16040818
127-
Abstract: PMID: 15539878
128-
Abstract: PMID: 10625187
129-
Abstract: PMID: 15909318
130-
Abstract: PMID: 7658512
131-
Abstract: PMID: 19888545
132-
Abstract: PMID: 1974458
133-
Abstract: PMID: 9718663
134-
Abstract: PMID: 21321968
135Rumack CM, Wilson, SR, and Charboneau JW. Eds. Diagnostic Ultrasound, 2nd edition, Mosby, 1998, pg.1100.
Above. 25 weeks gestation. Posterior urethral valves (PUV) with “keyhole” appearance to the bladder outlet and thickened bladder wall.
Above. Same patient, PUV. Sagittal view demonstrating dilated ureter.
Above. Same patient, PUV. Sagittal view of outlet and and thickened bladder wall.
Same patient, PUV. Oblique view with interface identified between the ureter and thickened bladder wall.
Same patient, PUV. Enlarged dysplastic kidney secondary to PUV outlet obstruction.
Above. Same patient (25 weeks gestation), PUV. Transverse view of the thickened bladder wall following aspiration of fluid from the fetal bladder.
Above. Patient B with PUV. Sagittal view with keyhole appearance of bladder outlet and dilated ureter.
Above. Patient B, PUV. Bladder is more distended and outlet obstruction is more pronounced.
Above. Patient B, PUV. Transverse view demonstrating thickened bladder wall and ureteral dilatation.
10 year survival: 82%
Kidney length greater than 97th percentile: 92%
Systemic hypertension: 75%
Congenital hepatic fibrosis and portal hypertension: 44%
Overall molecular detection rate: 76.6%.
2. Neonatal ventilation is a predictor of mortality but those infants who survive the neonatal period have a better prognosis than originally thought.
3. Disease progress is organ specific between kidney and liver.
4 Only a subset of patients develop periportal hypertension.
2. Syndromes which are distinguished by features other than echogenic kidneys:
Above. Patient A. Bilateral enlarged kidneys with small cysts and anhydramnios.
Above. Patient A. Markedly enlarged kidney with small cysts and anhydramnios.
Patient B. Massively enlarged kidneys with small cysts, small thorax and absent amniotic fluid.
Above. Patient C. 32 2/7 weeks gestation. Bilateral echogenic kidneys with small cysts and oligohydramnios. The renal size is small. Differential diagnosis would include certain syndromes. (See Imaging Considerations, ARPKD).
Above. Patient C. 32 2/7 weeks gestation. Bilateral echogenic kidneys with small cysts and oligohydramnios. Outcome likely lethal.
Above. Patient C. 32 2/7 weeks gestation. Transverse. Bilateral echogenic kidneys with small cysts and oligohydramnios.
Above. Patient C. 32 2/7 weeks gestation. Dolicocephaly and oligohydramnios.
Above Patient D. Larger multiple cysts and enlarged kidneys bilaterally. Differential diagnosis includes bilateral MCDK.
Above Patient D. Larger multiple cysts and enlarged kidneys bilaterally. Outcome, likely lethal.
Above Patient D. Larger multiple cysts and enlarged kidneys bilaterally.
Cystic Renal Dysplasia
Above. Patient A. Sagittal view. Multiple cysts and echogenic parenchyma are noted. These findings are also seen in multi-cystic dysplastic kidney and the two are difficult to distinguish.
Patient B. Again cysts and echogenic tissue are observed.
Above. Patient C. This dysplastic echogenic kidney’s findings are secondary to posterior urethral valves.
(B) ureterocele with total duplication and intravesical development (10%);
(C) ureterocele with total duplication and extravesical development (62%); and
(D) ureterocele with ectopic ureter (3%).
a. Observation (47.2%)
b. Ureterocele puncture (35.6)
c. 16.3% chose partial nephrectomy (16.3%).
Above. Duplicated collecting system (DCS), 29 weeks gestation. Sagittal view. Note upper pole and lower pole with greater dilatation of the upper pole.
Above. Same patient, DCS. Transverse view with left kidney suggesting DCS with potential upper and lower poles.
Same patient, DCS. Sagittal view of contralateral (right) kidney, which is normal.
Above. Same patient, DCS. Note dilated ureter exiting from upper pole.
Above. Same patient, DCS. Similarly, less dilated ureter exits from lower pole.
Above. Same patient, DCS. Note fetal bladder and classic appearance of ureterocle, likely representing the distal end of the ureter exiting from the upper pole.
Above. Same patient, DCS. Transverse to oblique view of ureterocele.
Above. Patient B, DCS. 32 weeks gestation. Note dilated upper and lower poles of the kidney.
Patient B, DCS. Classic view of ureterocele suggesting the presence of DCS.
Patient B, DCS. Similar view of ureterocele suggesting the presence of DCS.
Umbilical arteries around the fetal bladder.
Renal Agenesis
If the fetus is in breech presentation, transvaginal ultrasound may be useful.
Above: 25 3/7 weeks gestation. Unilateral renal agenesis with dilated renal pelvis on the right. Left renal fossa area, probable adrenal gland. Amniotic fluid volume is normal.Above. Same patient. Unilateral renal agenesis. Coronal view with right kidney and left renal fossa seen with probable adrenal gland.Above. Same patient Unilateral renal agenesis. Color Doppler demonstrates present of right renal artery only.Above: A. 19 1/7 weeks gestation. Coronal view. Empty renal fossa with adrenal gland seen. Virtually no amniotic fluid identified.
Above: same patient. Color Doppler demonstrates the absence of renal arteries bilaterally.
Above: same patient. Color power Doppler demonstrates the absence of renal arteries bilaterally.
Above B. 18 3/7 weeks gestation. Bilateral umbilical arteries are seen in their normal position but there is no fluid within the bladder.
Above. Same patient B. Again, empty renal fossa with adrenal gland visualized.
Above C. 22 6/7 weeks gestation. Transverse view. Bilateral empty renal fossae with a portion of the adrenal seen. Amniotic fluid is absent.
Above C. 22 6/7 weeks gestation. Color power Doppler demonstrating absent renal arteries. Coronal view with empty renal fossae.
Horseshoe Kidney, Pelvic Kidney, Crossed Fused Renal Ectopia
Above are sonographic features and findings specific to the antenatal diagnosis of horseshoe kidney. [128]
Horseshoe Kidney
Above. Horsehoe kidney. Coronal view. Elements of renal tissue join in the mid-line.
Above. Same patient. Coronal view. Definitive renal tissue can be seen in the mid-line (isthmus).
Above. Kidneys joining in the mid-line with definitive “horseshoe” shape.
Above: 20.1 weeks gestation. Unilateral renal agenesis with pelvic kidney. Echogenic tissue immediately inferior to empty fossa likely adrenal gland.
Above. Same patient demonstrating color power Doppler blood flow with a single renal artery to the pelvic kidney.
Same patient. Transverse view of the pelvis at the level of the bladder demonstrating pelvic kidney adjacent to the bladder.
Same patient. Transverse view of the pelvis at the level of the bladder demonstrating ureter and the pelvic kidney adjacent to the bladder.
Above: 29 5/7 weeks gestation. Right pelvic kidney adjacent to the bladder with cystic structure, possible ureterocele.
Above. Patient with enlarged kidney on the left side extending to the pelvis and solitary kidney on the right side.
Above. Same patient. Color flow Doppler indicates two renal arteries to the left kidney and one renal artery to the right kidney. The left kidneys extend to the plevis. A renal pelvis is identified in two segments on the left kidney.
Above. 29 weeks gestation. Renal ectopia with enlarged kidney extending to the pelvis; the contralateral kidney is absent.
| Abstract: PMID: 17602414 |
| Abstract: PMID: 21839591 |
| Abstract: PMID: 19838598 |
| Abstract: PMID: 21860792 |
| Abstract: PMID: 19237812 |
| Abstract: PMID: 15604570 |
| Abstract: PMID: 22046789 |
| Abstract: PMID: 18710723 |
| Abstract: PMID: 16515962 |
| Abstract: PMID: 19838598 |
| Abstract: PMID: 21413041 |
| Abstract: PMID: 16817090 |
| Abstract: PMID: 14691987 |
| Abstract: PMID: 15558286 |
| Abstract: PMID: 19838598 |
| Abstract: PMID: 15126863 |
| Abstract: PMID: 21748651 |
| Abstract: PMID: 16094041 |
| Abstract: PMID: 19373494 |
| Abstract: PMID: 19833558 |
| Abstract: PMID: 19536081 |
| Abstract: PMID: 19838598 |
| Abstract: PMID: 15470205 |
| Abstract: PMID: 16006956 |
| Abstract: PMID: 15912376 |
| Abstract: PMID: 19623154 |
| Abstract: PMID: 19838598 |
| Abstract: PMID: 15604570 |
| Abstract: PMID: 22046789 |
| Abstract: PMID: 18710723 |
| Abstract: PMID: 19838598 |
| Abstract: PMID: 14741187 |
| Abstract: PMID: 10835131 |
| Abstract: PMID: 3153330 |
| Abstract: PMID: 16174570 |
| Abstract: PMID: 1984303 |
37
| Boutheina BR, Aïda M, Lamia S, Ali M, MedBadis C, Samy J, Issam L, Ezzeddine S, et al. Lethal uropathies: prenatal diagnosis and feto-pathologic aspects. Tunis Med. 2000 Feb;78(2):120-4.
Abstract: PMID: 10894048 | |
| Abstract: PMID: 15108277 |
| Abstract: PMID: 9511976 |
| Abstract: PMID: 15698423 |
| Abstract: PMID: 2453819 |
| Abstract: PMID: 15164404 |
| Abstract: PMID: 15108281 |
| Abstract: PMID: 15698423 |
| Abstract: PMID: 12728091 |
| Abstract: PMID: 1781072 |
| Abstract: PMID: 18596710 |
| Abstract: PMID: 9166801 |
| Abstract: PMID: 8559760 |
| Abstract: PMID: 1606120 |
| Abstract: PMID: 1536230 |
| Abstract: PMID: 10401236 |
| Abstract: PMID: 1785280 |
| Abstract: PMID: 2204162 |
| Abstract: PMID: 3287366 |
| Abstract: PMID: 12504976 |
| Abstract: PMID: 11770813 |
| Abstract: PMID: 16032764 |
59
| Khoory BJ, Fanos V. Renal polycystosis in pediatrics. Pediatr Med Chir. 1998 Jul-Aug;20(4):269-73.
Abstract: PMID: 9866850 | |
| Abstract: PMID: 16491512 |
| Abstract: PMID: 14975971 |
| Abstract: PMID: 9564103 |
| Abstract: PMID: 9197441 |
| Abstract: PMID: 8542471 |
| Abstract: PMID: 17094077 |
| Abstract: PMID: 1870925 |
| Abstract: PMID: 1870925 |
| Abstract: PMID: 12386283 |
| Abstract: PMID: 12386283 |
| Abstract: PMID: 11685705 |
| Abstract: PMID: 11168926 |
72
| Shimada K, Hosokawa S, Matsumoto F, Matsumoto S. Pathological study of the kidney and the lung in patients with Potter sequence. Nihon Hinyokika Gakkai Zasshi. 1997 Jul;88(7):664-9.
Abstract: PMID: 9267130 | |
| Abstract: PMID: 6393764 |
| Abstract: PMID: 8518103 |
| Abstract: PMID: 12432113 |
76
| http://en.wikipedia.org/wiki/Duplicated_ureter | |
| Abstract: PMID: 6502807 |
| Abstract: PMID: 1000896 |
| Abstract: PMID: 21994077 |
| Abstract: PMID: 3492875 |
| Abstract: PMID: 20854476 |
| Abstract: PMID: 14734907 |
| Abstract: PMID: 22121240 |
| Abstract: PMID: 20728105 |
| Abstract: PMID: 21168903 |
| Abstract: PMID: 20728127 |
| Abstract: PMID: 15758799 |
| Abstract: PMID: 22398644 |
| Abstract: PMID: 15821573 |
| Abstract: PMID: 21527211 |
| Abstract: PMID: 21256545 |
| Abstract: PMID: 21437700 |
| Abstract: PMID: 409121 |
| Abstract: PMID: 14676444 |
| Abstract: PMID: 8705408 |
| Abstract: PMID: 10400048 |
| Abstract: PMID: 3492875 |
| Abstract: PMID: 21632996 |
| Abstract: PMID: 12704741 |
| Abstract: PMID: 12386558 |
| Abstract: PMID: 21897576 |
| Abstract: PMID: 8540439 |
| Abstract: PMID: 8201519 |
| Abstract: PMID: 20811621 |
| Abstract: PMID: 9037946 |
| Abstract: PMID: 21536809 |
| Abstract: PMID: 16116932 |
108
| O’Rahilly R, Muller, F. Human Embryology and Teratology, Wiley Liss, NY, 1996, pp. 279-279. | |
| Abstract: PMID: 16174570 |
| Abstract: PMID: 7877959 |
| Abstract: PMID: 16977473 |
| Abstract: PMID: 6393764 |
| Abstract: PMID: 20811621 |
| Abstract: PMID: 18835865 |
| Abstract: PMID: 1814304 |
| Abstract: PMID: 20811621 |
| Abstract: PMID: 8540439 |
| Abstract: PMID: 16053904 |
| Abstract: PMID: 22581658 |
| Abstract: PMID: 8540439 |
| Abstract: PMID: 22581658 |
| Abstract: PMID: 20618240 |
123
| Abourachid H, Dahmani F, Louis D, Hakami F, Daher N. Etiology of stenoses of the urethra. II–After trans-vesical prostatectomy. Apropos of 2 comparative series. Review of the literature. J Urol (Paris) 1989;95(7):412-4.Abstract: PMID: 2687395 | |
| Abstract: PMID: 21084958 |
125
| Markov D, Atanassova D, Pavlova E, Markov P. Empty renal fossa–a prenatal diagnostic dilemma. Akush Ginekol (Sofiia). 2010;49(5):13-9.
Abstract: PMID: 21268397 | |
| Abstract: PMID: 16040818 |
| Abstract: PMID: 15539878 |
| Abstract: PMID: 10625187 |
| Abstract: PMID: 15909318 |
| Abstract: PMID: 7658512 |
| Abstract: PMID: 19888545 |
| Abstract: PMID: 1974458 |
| Abstract: PMID: 9718663 |
| Abstract: PMID: 21321968 |
135
| Rumack CM, Wilson, SR, and Charboneau JW. Eds. Diagnostic Ultrasound, 2nd edition, Mosby, 1998, pg.1100. | |